Our main strategy for quantitative mass spectrometry based proteomics is a label-free approach using nanoUPLC-MSE analysis. Analyzing samples in MSE mode enables us to quantitatively compare hundreds of proteins across dozens of samples in a single experiment. These information-rich studies enabled us to identify novel biomarkers in serum, plasma, cerebrospinal fluid and tissues and to potentially identify disease subgroups.
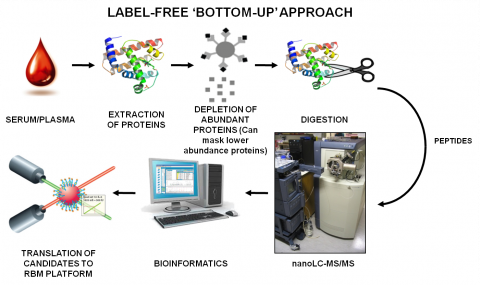
Figure: Shotgun mass spectrometry. 1) Proteins are digested with an enzyme to produce peptides, 2) the peptides are separated via HPLC, 3) the peptides are ionized by electrospray and 4) intact peptide ion intensities are used to calculate abundance and time-of-flight analysis is used to measure mass/charge ratios of the peptides and peptide fragments for identification via database comparisons
The mass spectrometer measures the mass-to-charge (m/z) ratio of the ionized peptides and peptide fragments, and peptides can then be identified by comparison of the m/z values with those in a protein database which has been subjected to virtual proteolysis. The main advantage of this method is the ability to detect more difficult classes of proteins which are not detectable by traditionally-used two-dimensional gel electrophoresis approaches.